The chemistry of compounds with a conjugated bond system is a developing research line in the field of synthesis of new organic materials. The properties of conjugated compounds are determined by electronic structure of their molecules and conjugation between the molecular fragments. Molecular systems with conjugated bonds and charge-transfer complexes are characterized by an electrical conductivity comparable to the conductivity of metals provided that their crystal structure creates favorable conditions for electron delocalization [1, 2].
Due to their peculiar properties, compounds with a conjugated bond system are in some cases capable of inhibiting chain reactions, including oxidation pro- cesses. Therefore, they can be used as polymer stabi- lizers under aging conditions. Unlike other stabilizers, in particular antioxidants possessing labile hydrogen atoms, conjugated compounds hamper chain transfer in the polymer decomposition process [3]. The use of compounds with a conjugated bond system, such as polyhydroquinones and polyquinones, as chemical stabilizers provides additional possibilities for the solution of problems related to elevated temperature limit of polymer processing and operation.
A number of methods for the synthesis of poly- hydroquinones and polyquinones have been reported; they include electrooxidative polymerization [4–6], enzymatic polymerization [7], and dehalogenation- based organometallic polycondensation [8]. The sim- plest procedure is based on chemical oxidative poly- merization [9–11].
The reduction of 3,3′,5,5′-tetra-tert-butyl-1,1′-bis- (cyclohexa-2,5-dien-1-ylidene)-4,4′-dione in the pres- ence of hydroquinone gives 3,3′,5,5′-tetra-tert-butylbi- phenyl-4,4′-diol and an oligomer (polyquinone) [12]. In this work we studied oxidative dehydrogenation of pyrocatechol (2a), hydroquinone (2b), and 1,4-benzo- quinone (2c) using 3,3′,5,5′-tetra-tert-butyl-trans-stil- benequinone [1, 4,4′-(ethane-1,2-diylidene)bis(2,6-di- tert-butylcyclohexa-2,5-dien-1-one)] as oxidant (Scheme 1). These reactions make it possible to obtain polyphenols 3a and 3b and polyquinones 3c and 3d together with 4,4′-(ethene-1,2-diyl)bis(2,6-di-tert- butylphenol) (4). The reactions were carried out in an aprotic solvent (toluene) in the presence of aqueous acid or alkali in the temperature range from 50 to 200°C (reaction time 30 min). Polyquinones 3c and 3d were separated from the aqueous phase by filtration,and compound 4 crystallized from the organic phase on cooling to room temperature. The reduction of stilbenequinone 1 to bis-phenol 4 requires two hydro- gen atoms; therefore, the reactants were taken in equimolar amounts.
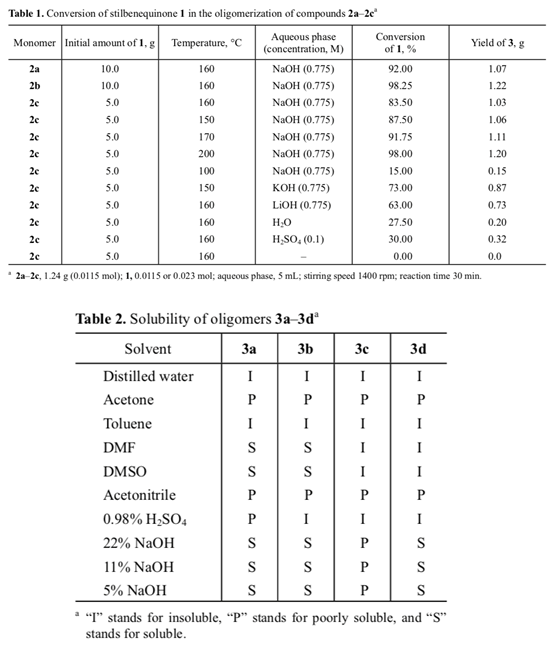
Unlike initial quinone 1, the IR spectrum of 4 (Fig. 1) contained no absorption band at 1605 cm–1 typical of the conjugated diene system and carbonyl group, but bands at 3627 and 1231–1133 (OH) and 1437 and 660–730 cm–1 (C=C) appeared. The melting point of bis-phenol 4 (mp 240°C) coincided with that reported in [13].
The proposed procedure is advantageous due to high conversion of stilbenequinone 1 (98%), high yield of the polyquinone (97%), and facile isolation of the products. Furthermore, initial stilbenequinone 1 can be readily regenerated from bis-phenol 4. Therefore, the described reaction may underlie a continuous process for the synthesis of polyquinones with a minimum consumption of stilbenequinone 1.
We examined the effects of temperature, acidity of the medium, concentration of alkali solution (Table 1), and solubility of the reactants in organic and inorganic solvents (Table 2) on the conversion of stilbene- quinone 1. The maximum conversion was reached when the reaction was carried out in the presence of aqueous sodium hydroxide. The conversion of 1 increased as the temperature rose and attained 98% at 200°C (Table 1). No reaction occurred in the absence of water, which indicated participation of the latter in the process [14].
Oligomers 3a–3d are insoluble in water and toluene and poorly soluble in acetone and acetonitrile. Poly- hydroquinone 3b and polypyrocatechol 3a are readily soluble in DMF and DMSO. All oligomers, except for 3c, are readily soluble in aqueous alkalies (Table 2).
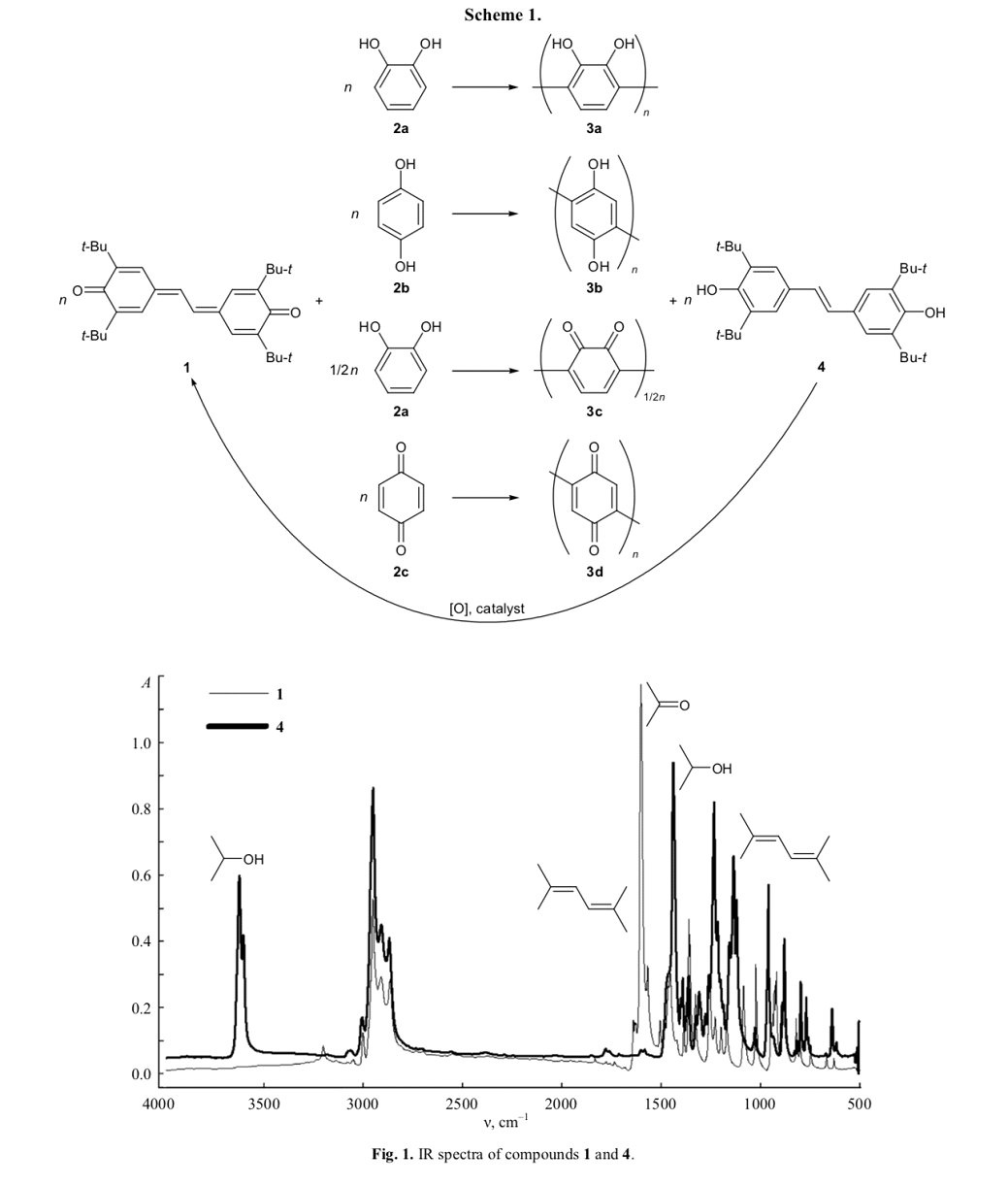
The molecular weights of 3a and 3b dissolved in DMF were determined by MALDI TOF mass spec- trometry. As follows from Figs. 2 and 3, these oligomers are represented by a set of species with different molecular weights, the largest of which being 1156.3 (3b) and 1421.4 (3a). The degree of polymer- ization ranges from 5 to 15 units.
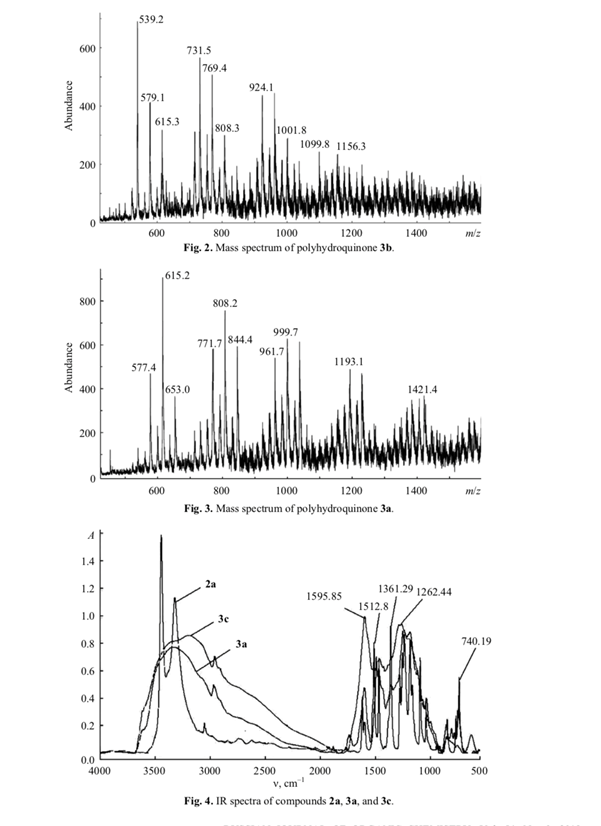
The IR spectra of 3a and 3c showed no absorption bands at 1361, 1100, and 675 cm–1, which were observed in the spectrum of 2a (Fig. 4). The presence of carbonyl groups in 3c followed from the appearance of IR band at 1595 cm–1. Broadened absorption bands in the region 3500–3000 cm–1 of the spectra of 3a and 3c indicated their oligomeric nature, and the band at 3200 cm–1 was assigned to polyassociates formed by these oligomers [4].
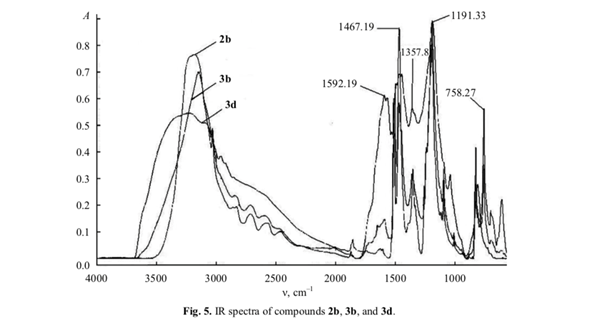
Among compounds 2b, 3b, and 3d, only the former displayed IR absorption bands at 610, 828, and 1855 cm–1 due to vibrations of aromatic C–H and C=C bonds (Fig. 5), whereas carbonyl stretching band was observed only in the spectrum of 3d at 1592 cm–1. The other bands in the spectra of 3b and 3d were consistent with their structure. The band at 3400 cm–1 appeared due to formation of polyassociates [4]. The elemental composition of 3d coincided with that calculated for (C6H2O2)n, which suggests the presence of identical functional groups in molecules 3d and 2c (C6H4O2).
EXPERIMENTAL
The IR spectra were recorded on a Perkin Elmer Spectrum Two spectrometer with Fourier transform. The 1H NMR spectrum of 1 was recorded on a Bruker Avance-600 instrument using DMSO-d6 as solvent and
reference (δ 2.50 ppm). The molecular weights of 3a and 3b were determined with a Bruker Daltonik MALDI TOF high-resolution mass spectrometer using 2,5-dihydroxybenzoic acid as matrix. The elemental analyses were obtained on a Carl Zeiss AURIGA Cross Beam workstation equipped with an Oxford Instru- ments INCAX-MAX energy-dispersive spectrometer (resolution 127 eV). The kinetic curves for the trans- formation of stilbenequinone 1 to bis-phenol 4 were plotted from the spectrophotometric data obtained on an Ekros PE5300V spectrophotometer at λ 520 nm. The reaction mixtures were analyzed by GLC on a Khromatek Kristall-5000 gas chromatograph.
The reactions were carried out in a 150-mL cylin- drical stainless steel reactor equipped with a pressure gage, thermocouple, temperature control probe, and a Heildolph MR–Hei–Standard magnetic stirrer with a heater and variable stirring speed. The reactor was charged with 5.0 g (0.0115 mol) of stilbenequinone 1, 1.24 g (0.0115 mol) of compound 2a–2c, an aqueous solution of sodium, lithium, or potassium hydroxide, and 50.0 mL of toluene. In the synthesis of 3c the amount of stilbenequinone 1 was twice as large. The reactions in the presence of aqueous sulfuric acid and without water were carried out in a similar way. The mixture was heated to 100–200°C, stirring was turned on, and the mixture was stirred for 30 min. The conversion of stilbenequinone 1 was determined by photocolorimetry at λ 520 nm. When the reaction was complete, stirring and heating were turned off. The reactor was opened at 85°C, and oligomeric product 3a–3d was filtered off (Table 1). The filtrate was cooled to room temperature, and compound 4 pre- cipitated. The bottom layer of the reaction mixture (aqueous alkaline solution) was neutralized to pH 7 with 0.1 N aqueous HCl, and the black solid was fil- tered off and washed with distilled water.
4,4′-(Ethane-1,2-diylidene)bis(2,6-di-tert-butyl- cyclohexa-2,5-dien-1-one) (1). Reaction temperature 90°C, reaction time 90 min, oxygen flow rate 300 h–1, 10% sodium hydroxide, solvent kerosene (50 mL), stirring speed 1400 rpm [15]. Yield 99%, mp 315°C. IR spectrum, ν, cm–1: 3003 (C–Harom), 2952, 2909, 2865 (Me), 1640, 1605 (C=Carom), 1605 (C=O), 1600, 1454 (C=Carom), 1360, 1256 (t-Bu). 1H NMR spectrum (DMSO-d6, 600 MHz), δ, ppm: 1.42 s (36H, t-Bu), 6.54 s (2H, =CH), 7.19 s (4H, Harom). Found, %: C 82.80, 82.56; H 9.73. C30H42O2. Calculated, %: C 82.55; H 10.15.
Polypyrocatechol (3a). Yield 86%. IR spectrum, ν, cm–1: 3600 (OH), 1600, 1512 (C=Carom), 1350, 1250, 1180 (C–OH).
Polyhydroquinone (3b). Yield 89%. IR spectrum, ν, cm–1: 3150 (OH), 1590, 1510 (C=C ) 1352, 1350, 1185 (C–OH).
REFERENCES
1. Tibaoui, T., Ayachi, S., Hamidi, M., Bouachrine, M., Paris, M., and Alimi, K., J. Appl. Polym. Sci., 2010, vol. 118, p. 711.
2. Akhmadullin, R.M., Akhmadullina, A.G., Mukmene- va, N.A., Gazizov, A.R., Tagirov, L.R., Klochkov, V.V., Yavkin, B.V., Orlinskii, S.B., and Nigmatullin, T.F., Res. J. Pharm. Biol. Chem. Sci., 2016, vol. 7, no. 2, p. 577.
3. Furlani, A., Russo, M.V., and Cataldo, F., Synth. Met., 1989, vol. 29, no. 1, p. 507.
4. Sadykh-Zade, S.I., Ragimov, A.V., Suleimanova, S.S., and Liogon’kii, B.I., Vysokomol. Soedin., 1972, vol. 14, no. 6, p. 1395.
5. Zhang, A.J., He, J., Guan, Y., Li, Z.Y., Zhang, Y.J., and Zhu, J.X., Sci. China: Chem., 2012, vol. 55, p. 5.
6. Yamamoto, K., Asada, T., Nishide, H., and Tsushida, E., Bull. Chem. Soc. Jpn., 1990, vol. 63, no. 4, p. 1211.
7. Wang, P., Martin, B.D., Parida, S., Rethwisch, D.G., and Dordick, J.S., J. Am. Chem. Soc., 1995, vol. 117, no. 51, p. 12885.
8. Berlin, A.A., Ragimov, A.V., Sadykh-Zade, S.I., Gadzhieva, T.A., and Takhmazov, B.M., Vysokomol. Soedin., 1975, vol. 17, no. 1, p. 111.
9. Yamamoto, T. and Kimura, T., Macromolecules, 2011, vol. 44, no. 7, p. 2245.
10. Sabaa, M.W., Madkour, T.M., and Yassin, A.A., Polym. Degrad. Stab., 1988, vol. 22, p. 195.
11. Akhmadullin, R.M. and Akhmadullina, A.G., RU Patent no. 2552516, 2015.
12. Akhmadullin, R.M., Gatiyatullin, D.R., Akhmadulli- na, A.G., Verizhnikov, L.V., and Mukmeneva, N.A., Vestn. Kazan. Tekhnol. Univ., 2014, vol. 17, no. 6, p. 160.
13. Bonn, C.R. and Compbell, T.W., J. Org. Chem., 1957, vol. 22, no. 4, p. 458.
14. Ragimov, A.V., Bektashi, F.T., and Liogon’kii, B.I., Vysokomol. Soedin., 1975, vol. 17, no. 12, p. 2753.
15. Hoang, H.Y., Akhmadullin, R.M., Akhmadullina, F.Y., Zakirov, R.K., Akhmadullina, A.G., and Gazizov, A.S., Russ. J. Org. Chem., 2018, vol. 54, p. 1008.