An important environmental problem in oil proces- sing and petrochemical industries is efficient neutral- ization of sulfide-containing alkaline wastes before their discharge into biological treatment facilities. For this purpose, various methods of catalytic liquid-phase oxidation with atmospheric oxygen are widely used in industry [1–4]. The main disadvantages of the existing catalysts include low catalytic activity, low selectivity, and low stability in alkaline medium. In this work we have synthesized 3,3′,5,5′-tetra-tert-butyl-4,4′-stilbene- quinone [1, IUPAC name 4,4′-(ethane-1,2-diylidene)- bis(2,6-di-tert-butylcyclohexa-2,5-dien-1-one)], con- firmed its structure, and studied its catalytic activity in the liquid-phase oxidation of Na2S with oxygen.
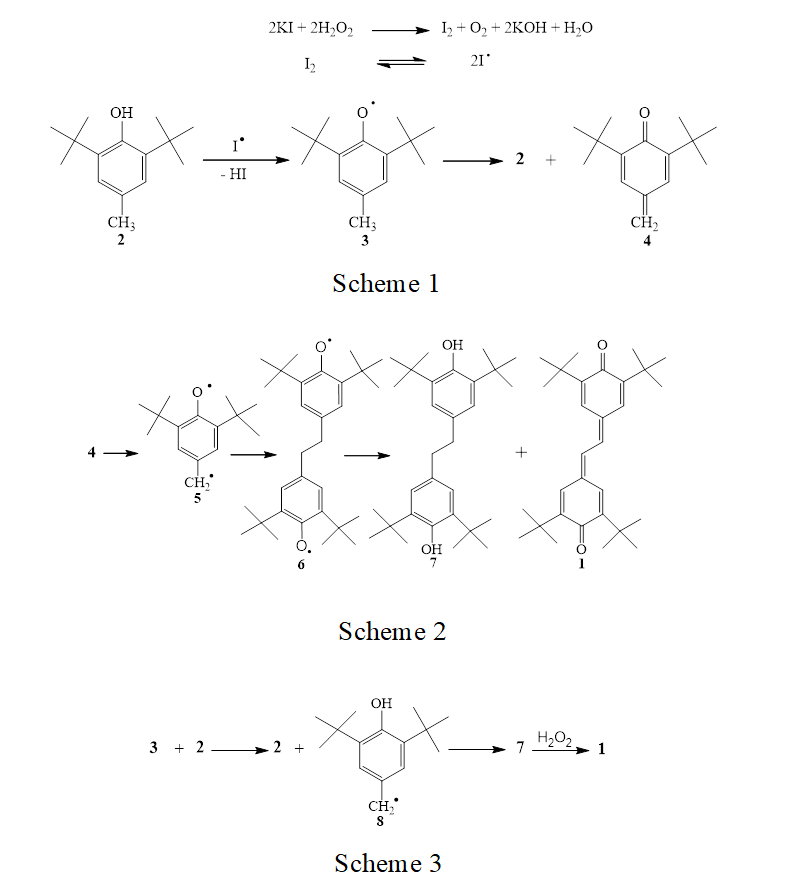
Stilbenequinone 1 was synthesized by oxidation of 2,6-di-tert-butyl-4-methylphenol (2) with hydrogen peroxide in the presence of potassium iodide. The formation of stilbenequinone 1 is the result of C–C dimerization of phenoxy radicals generated by oxida- tion of sterically hindered phenol 2, which was ac- companied by dehydrogenation [5]. The formation of phenoxy radicals in the oxidation of 2 was confirmed by ESR and was thoroughly reviewed in [6–9]. The primary act of the oxidation is elimination of hydrogen from the hydroxy group [7, 10, 11] by the action of iodine atom arising from homolytic dissociation of molecular iodine (Scheme 1). Phenoxy radical 3 pos- sesses an alkyl group in the para position and is unstable. It undergoes disproportionation to initial phenol 2 and methylenequinone 4 [12, 13]. The reaction mixture acquires lemon yellow tint due to formation of 4. Spontaneous dimerization of unstable methylene- quinone 4 gives bisphenoxy radical 6 which is con- verted to a mixture of stilbenequinone 1 and dihy- droxydiphenylethane 7 (Scheme 2) [12–15].
Apart from disproportionation, initially formed radical 3 is capable of isomerizing to hydroxybenzyl radical 8 via hydrogen transfer from the methyl group to oxygen [16–21]. Dimerization of 8 leads to the formation of compound 7 which is oxidized with hy- drogen peroxide to final stilbenequinone 1 (Scheme 3).
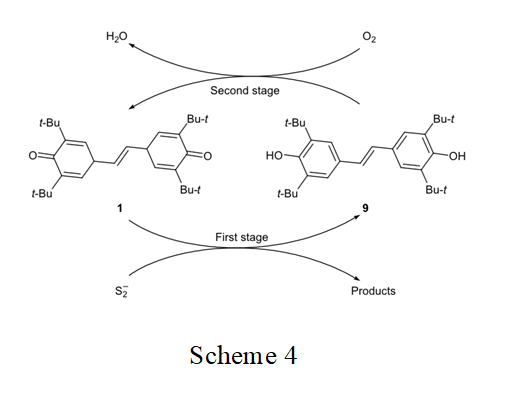
In the preceding study [22] we presumed that stilbenequinone acts as redox catalyst in the oxidation of sulfide sulfur. This process is likely to include two stages: (1) oxidation of S2– with stilbenequinone 1 and reduction of the latter to dihydroxydiphenylethene 9 and (2) regeneration of the catalyst by oxidation of 9 in alkaline medium (Scheme 4). In order to confirm the proposed mechanism, in this work we studied the oxidation of sodium sulfide with stilbenequinone 1 at appeared (960 cm–1). The melting point of the product coincided with that reported in the literature for com- pound 9 (240°C [21]).
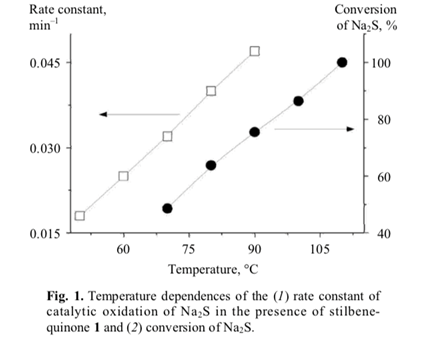
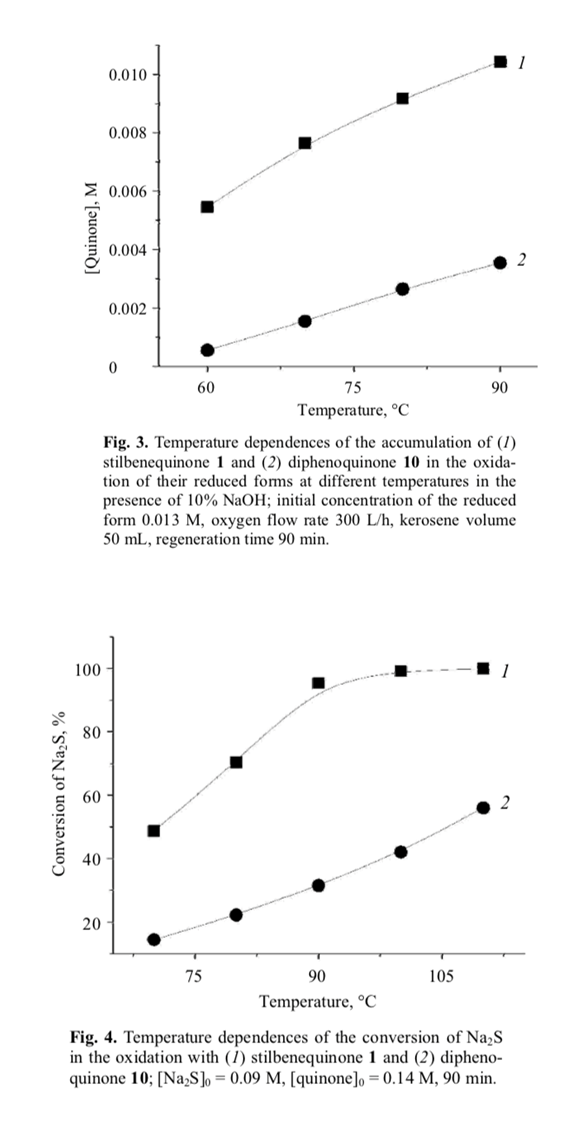
The catalytic activity of stilbenequinone 1 is determined by a number of factors, including temperature, pH, sulfide nature, etc. The rate of oxidation of sulfide sulfur (Fig. 1) and regeneration of stilbenequinone 1 (Fig. 3) increased as the temperature rose; correspond- ingly, the efficiency of catalytic oxidation increased.
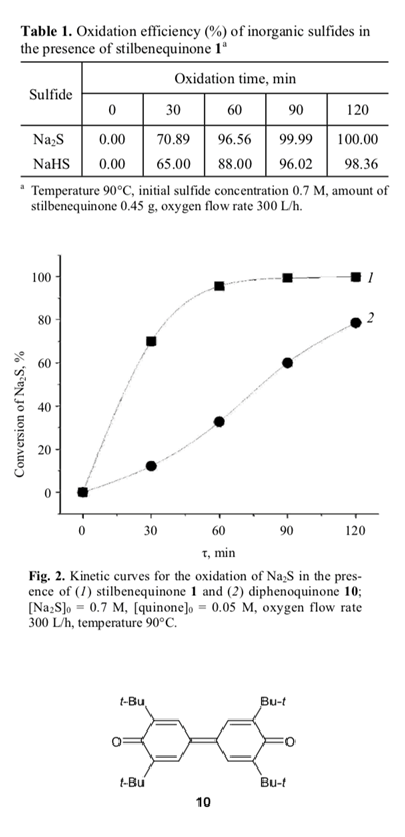
The catalytic activity of stilbenequinone is also sensitive to pH of the medium. The data in Table 1 illustrate variation of the conversion of sodium sulfide (pH 13.5) and sodium hydrogen sulfide (pH 9) in the presence of stilbenequinone 1. It is seen that the rate of oxidation of Na2S is higher than the rate of oxidation of NaHS. According to published data [8, 23], hydroxide ions catalyze oxidation of hydroquinone to benzoquinone; therefore, higher concentration of OH– ions favors regeneration of the catalyst in the oxidation of sulfide sulfur.
Addition of an inhibitor could reduce the catalytic activity or completely deactivate the catalyst. This was observed in the oxidation of ammonium sulfide in the presence of stilbenequinone 1. The rate of catalytic oxidation of (NH4)2S was the same as the rate of non- catalytic process. Furthermore, addition of aqueous ammonia to a solution of Na2S appreciably reduced the rate of catalytic oxidation of the latter: y = –0.0098x + 0.0111, where y is the rate of catalytic oxidation of Na2S, and x is the concentration of ammonia.
The observed pattern can be rationalized as follows. Quinonesareconjugatedα,β-unsaturateddicarbonyl compounds characterized by large dipole moments, and excess electron density therein is localized on the oxygen atom. Therefore, quinones are very sensitive to electrophiles [24]. Ammonium ions generated by dis- sociation of ammonium sulfide are strong electrophilic species capable of blocking active sites of the catalyst with subsequent complete deactivation.
The structure of quinones largely determines their ability to participate in chemical reactions. With the goal of elucidating the relation between the structure of quinones and their catalytic activity in the oxidation of sodium sulfide, we compared the activities of stilbene- quinone 1 and 3,3′,5,5′-tetra-tert-butyl-4,4′-dipheno- quinone [1,1′-bi(cyclohexa-2,5-dien-1-ylidene)-4,4′-di- one] 10. It is seen (Fig. 2) that the catalytic activity of 1 is higher than the activity of 10. Presumably, this is related to the mechanism of oxidation of sulfide sulfur in the presence of quinones, which is determined by both stages (Scheme 4).
This assumption was confirmed by the results of oxidation of Na2S with quinones in an inert medium (first stage in Scheme 4). Stilbenequinone 1 is superior to diphenoquinone 10 in the oxidizing ability (Fig. 4). Unlike compound 10, the two cyclohexadiene frag- ment in molecule 1 are separated bu two CH units. The higher reactivity of stilbenequinone 1 is determined by higher accessibility of the CH groups to attack of nucleophile [25, 26].
The second stage in the catalytic oxidation of sulfide sulfur in the presence of quinones (Scheme 4) is the oxidation of 9 or 3,3′,5,5′-tetra-tert-butyl-1,1′-bi- phenyl-4,4′-diol (11, reduced form of 10). Here, the polarity of the hydroxy group is important. Change of the O–H bond polarity in going from one hydro- quinone to another determines the difference in the energies of dissociation of that bond and hence their different oxidizabilities [6].
Unlike compound 11, the conjugated bond system in molecule 9 includes the CH=CH double bond in addition to the aromatic rings. Therefore, the electro- negativity of the oxygen atom in 9 is higher, and the Coulomb integral of the oxygen δ-orbitals is more negative. Such change of the δ-electron structure of the OH bond increases its polarity and reduces the energy of dissociation, so that compound 9 should be oxidized more readily than 11. This was confirmed experi- mentally (Fig. 4).
Thus, we have shown that the rate of regeneration of stilbenequinone 1 is higher than the rate of regen- eration of diphenoquinone 10.
EXPERIMENTAL
The IR spectra were recorded on a Perkin Elmer Spectrum Two spectrometer equipped with an ATR accessory. The 1H NMR spectra were recorded on a Bruker Avance-600 spectrometer using DMSO-d6 as solvent and reference (δ 2.50 ppm). The elemental analysis of stilbenequinone was obtained with an AURIGA Cross Beam workstation equipped with an IncaX-MAX energy dispersive spectrometer (reso- lution 127 eV). The melting points were measured on a Buchi M-560 melting point apparatus.
The concentration of quinone was determined by photocolorimetry. A 0.5-cm3 sample of kerosene frac- tion was withdrawn from a three-necked cylindrical reactor without preliminary cooling. The sample was diluted with toluene to a volume of 50 cm3 (by a factor of 100) in a volumetric flask. The kerosene fraction containing dissolved quinone endows toluene solution with a bright yellow color which becomes more intense as the concentration of quinone increases. The absorbance at λ 500 nm was measured with an Ekros PE5300V spectrophotometer using a 10-cm path- length cell, and the concentration of quinone was cal- culated using a calibration curve.
The concentration of sulfides was determined by potentiometric titration according to GOST (state standard) no. 22985.
4,4′-(Ethane-1,2-diylidene)bis(2,6-di-tert-butyl- cyclohexa-2,5-dien-1-one) (1). A mixture of 30 g of compound 2 and 3 g of potassium iodide in 120 mL of propan-2-ol was stirred at 70°C in a 500-mL cylindrical glass reactor. After 30 min, 42 mL of a 35% aqueous solution of hydrogen peroxide was added, and the mixture was stirred for 9 h at 70–75°C. The mixture was cooled, and the red–orange crystal- line solid was filtered off, recrystallized from toluene, and dried for 1 h at 120°C. Yield 98%, mp 315°C. IR spectrum, ν, cm–1: 3003 (C–Harom), 2952, 2909, 2865 (Me), 1640, 1605 (=C–C=), 1605 (C=O), 1600, 1454 (C=Carom), 1360, 1256 (t-Bu). 1H NMR spectrum (600 MHz), δ, ppm: 1.42 s (36H, t-Bu), 6.54 s (2H, =CH), 7.19 s (4H, C6H2). Found, %: C 82.80, 82.56; H 9.73; O 7.47, 7.71. C30H42O2. Calculated, %: C 82.55; H 10.15; O 7.30.
Catalytic oxidation of inorganic sulfides was carried out in a 150-ml cylindrical glass reactor which was charged with a mixture of 40 mL of an inorganic sulfide solution, 20 mL of kerosene, and a required amount of the catalyst. Oxygen was passed through the mixture under stirring at 1400 rpm using a magnetic stirrer on heating at a required temperature.
The oxidation of inorganic sulfides with quinones in an inert medium was carried out in a 150-mL stain- less steel high-pressure reactor. A mixture of 40 mL of an inorganic sulfide solution, 20 mL of kerosene, and a required amount of the catalyst was stirred at 1400 rpm.
Hydrogen peroxide of chemically pure grade, 2,6-di-tert-butyl-4-methylphenol, propan-2-ol, potassium iodide, sodium sulfide, toluene, and aqueous ammonia of analytical grade, 3,3′,5,5′-tetra-tert-butyl- 4,4′-diphenoquinone, 3,3′,5,5′-tetra-tert-butyl-1,1′-bi- phenyl-4,4′-diol, and straight kerosene fraction of pure grade, and oxygen and argon of ultrapure grade (from gas cylinders) were used. Solutions of sodium hydro- gen sulfide and ammonium sulfide were prepared according to the procedures described in [27, 28].
REFERENCES
1. Akhmadullin, R.M., Bui, D.N., Akhmadullina, A.G., and Samuilov, Ya.D., Kinet. Catal., 2013, vol. 54, no. 3, p. 334.
2. Bui, D.N., Akhmadullin, R.M., Akhmadullina, A.G., and Aghajanian, S.I., J. Sulfur Chem., 2013, vol. 35, no. 1, p. 74.
3. Akhmadullin, R.M., Akhmadullina, A.G., and Agadzha- nyan, S.I., RU Patent no. 2529500, 2014.
4. Bui, D.N., Akhmadullin, R.M., Akhmadullina, A.G., Samuilov, Y.D., and Aghajanian, S.I., ChemPhysChem, 2013, vol. 14, no. 18, p. 4149.
5. Akhmadullin, R.M., Cand. Sci. (Chem.) Dissertation, Kazan, 2009.
6. Strigun, L.M., Vartanyan, L.S., and Emmanuel’, L.M., Russ. Chem. Rev., 1968, vol. 37, no. 6, p. 421.
7. Pokhodenko, V.D., Khizhnyi, V.A., and Bidzilya, V.A., Russ. Chem. Rev., 1968, vol. 37, no. 6, p. 435.
8. Shanina, E.L., Zaikov, G.E., and Mekmeneva, N.A., Polym. Degrad. Stab., 1996, vol. 51, p. 51.
9. Buchachenko, A.A., Stabil’nye radikaly (Stable Radicals), Moscow: Nauka, 1963.
10. Cook, C.D., Nash, N.G., and Flanagan, H.R., J. Am. Chem. Soc., 1955, vol. 77, p. 1783.
11. Pokhodenko, V.D., Ganyuk, L.N., Yakovleva, E.A., Shatenshtein, A.I., and Brodskii, A.I., Dokl. Akad. Nauk SSSR, 1963, vol. 148, no. 1, p. 1314.
12. Rozantsev, E.G. and Sholle, V.D., Organicheskaya khimiya svobodnykh radikalov (Organic Chemistry of Free Radicals), Moscow: Khimiya, 1979.
13. Reutov, O.A., Kurts, A.L., and Butin, K.P., Organi- cheskaya khimiya (Organic Chemistry), Moscow: Binom. Laboratoriya Znanii, 2004, vol. 3.
14. Bauer, R. and Coppinger, G., Tetrahedron, 1963, vol. 19, p. 1201.
15. Ershov, V.V. and Volod’kin, A.A., Izv. Akad. Nauk SSSR, Otd. Khim. Nauk, 1962, p. 1290.
16. Previc, E.P., Hotelling, E.B., and Neuworth, M.W., Ind. Eng. Chem., 1961, vol. 53, p. 469.
17. Bickel, A.F. and Kooyman, Ε.S., J. Chem. Soc., 1953, p. 3211.
18. Campbell, T.W. and Coppinger, G.M., J. Am. Chem. Soc., 1952, vol. 74, p. 1469.
19. Campbell, T.W. and Coppinger, G.M., J. Am. Chem. Soc., 1952, vol. 74, p. 1469. Emanuel’, N.M., Dronova, L.M., Konovalova, N.P., Maizus, Z.K., and Skabida, I.P., Dokl. Akad. Nauk SSSR, 1963, vol. 152, p. 481.
20. Konovalova, N.P., Bogdanov, G.N., Vasil’eva, Ya.S., Dronova, L.M., Matveeva, A.L., and Emanuel’, N.M., Dokl. Akad. Nauk SSSR, 1966, vol. 168, p. 1419.
21. Bohn, C.R. and Compbell, T.W., J. Org. Chem., 1957, vol. 22, no. 4, p. 458.
22. Kha, T.Z., Khoang, Kh.I., Akhmadullina, F.Yu., Akhma- dullin, R.M., Zakirov, R.K., and Akhmadullina, A.G., Voda: Khim. Ekol., 2017, vol. 5, p. 31.
23. Karasch, M.S. and Jochi, B.S., J. Org. Chem., 1957, vol. 22, no. 11, p. 1439.
24. Agronomov, A.E., Izbrannye glavy organicheskoi khimii: uchebnoe posobie dlya vuzov (Selected Topics of Organic Chemistry. Textbook for Higher Educational Institutions), Moscow: Khimiya, 1990.
25. Bukharov, S.V., Mukmeneva, N.A., and Nugumano- va, G.N., Vestn. Kazan. Tekhnol. Univ., 2012, vol. 5, no. 23, p. 94.
26. Volod’kin, A.A. and Ershov, V.V., Russ. Chem. Rev., 1988, vol. 57, no. 4, p. 336.
27. Manganaro, J.L., US Patent no. 4 439 411, 1984.
28. Karyakin, Yu.V. and Angelov, I.I., Chistye khimicheskie veshchestva (Pure Chemicals), Moscow: Khimiya, 1974.